Mudar a história: Como o sítio nitisinone devolve a esperança aos doentes com HT-1 e AKU
Última atualização: 15 de janeiro de 2024


Pode aceder legalmente a novos medicamentos, mesmo que estes não estejam aprovados no seu país.
Saiba comoQuando se é confrontado com uma doença rara, encontrar um incentivo pode parecer tão importante como encontrar um tratamento. Estamos aqui para o apoiar em ambos, começando com o exemplo da Tirosinemia hereditária de tipo 1 (HT-1) e da Alcaptonúria (AKU).
Apesar de a HT-1 e a AKU afectarem cerca de 1 em cada 100.000 pessoas, tratamentos como o nitisinone ajudaram a alterar o prognóstico dos doentes. E com os Programas de Acesso Precoce (especialmente na Índia, Paquistão, Bangladesh e Sudão), mais doentes podem aceder ao tratamento, independentemente da sua situação financeira.
Eis tudo o que precisa de saber sobre Nitisinone e como pode aceder-lhe.
O que é o HT-1?
A tirosinemia hereditária tipo 1 (HT-1) é uma doença genética rara que afecta o metabolismo de um aminoácido chamado tirosina. A tirosina está presente na maioria das proteínas.
Os doentes com HT-1 têm uma acumulação de resíduos de tirosina no organismo. Eventualmente, isto pode danificar o fígado e os rins, e aumentar o risco de cancro do fígado. Alguns doentes podem também sentir amolecimento e enfraquecimento dos ossos, bem como problemas no sistema nervoso [1, 2].
Quão raro é o HT-1?
A HT-1 afecta cerca de 1 em 100.000 pessoas em todo o mundo [3]. Curiosamente, em algumas regiões do Canadá, a incidência é consideravelmente mais elevada, afectando 1 em cada 1846 recém-nascidos [4]. Presume-se que este facto se deva a uma mutação fundadora na população franco-canadiana. Também se observa uma maior incidência de HT-1 na Turquia e na Índia [4].
A investigação entre diferentes populações no Reino Unido sugeriu que as pessoas de ascendência paquistanesa podem ser afectadas pelo HT-1 com mais frequência do que as de ascendência europeia (3,7 casos de HT-1 por milhão contra 0,04 casos por milhão, respetivamente) [5].
Sintomas de tirosinemia
Os sintomas da HT-1 podem aparecer em diferentes alturas da vida. Alguns doentes apresentam sintomas logo no primeiro ano de vida, enquanto noutros podem surgir anos mais tarde.
Os tipos de sintomas diferem consoante os doentes e podem incluir raquitismo, aumento anormal do tamanho do baço ou do fígado ou insuficiência hepática aguda [2].
Os primeiros sintomas da HT-1 podem ser difíceis de detetar. No entanto, iniciar o tratamento o mais cedo possível faz uma grande diferença nos resultados dos doentes. É por isso que o rastreio neonatal é importante [4].
Como é que o Nitisinone trata o HT-1?
Nitisinone facilita a degradação da tirosina. Este facto ajuda a prevenir a toxicidade hepática e renal, bem como os danos que provocam [7].
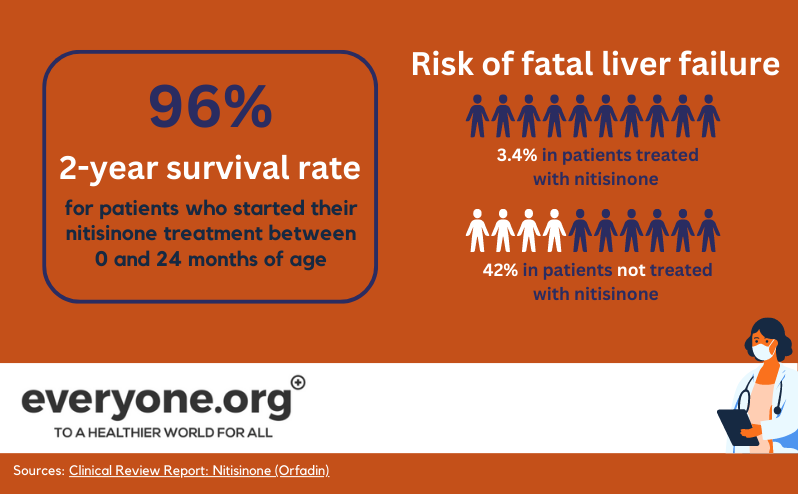
Antes da introdução de nitisinone, os doentes com HT-1 muitas vezes não chegavam a viver até à adolescência. A insuficiência hepática ou renal resultante da toxicidade seria fatal para muitos [6]. O transplante de fígado costumava ser a única forma de corrigir o metabolismo da tirosina dos doentes com HT-1.
Nitisinone Os comprimidos vieram alterar esta situação. Mais de 90% dos doentes respondem muito bem ao tratamento com nitisinone, em combinação com uma dieta pobre em proteínas. Como resultado do tratamento com nitisinone , atualmente só são necessários transplantes de fígado em casos raros [1]. Os doentes que iniciaram o tratamento precocemente vivem até à idade adulta e têm uma vida bastante normal [15].
Nitisinone como tratamento da alcaptonúria (AKU)
A alcaptonúria (também conhecida como doença dos ossos negros, AKU ou doença da urina negra) é uma doença hereditária rara que apresenta alguma semelhança com a HT-1. Os doentes com AKU apresentam perturbações no metabolismo da oxidase do ácido homogentísico (HGA) - uma enzima que participa no metabolismo de alguns aminoácidos, como a tirosina e a fenilalanina[11].
Uma vez que ambos partilham uma via metabólica semelhante, a adequação do nitisinone como tratamento da alcaptonúria também foi estudada [12].
De acordo com os resultados do estudo, o nitisinone parece ser um tratamento eficaz para a AKU em doentes adultos e uma prevenção eficaz para as complicações da AKU em crianças [12]. Também em doentes com AKU, o tratamento com nitisinone deve ser combinado com restrição dietética de proteínas, particularmente tirosina e fenilalanina [1].
São necessários mais estudos para confirmar a eficácia do nitisinone no tratamento da alcaptonúria, bem como para estabelecer quaisquer potenciais riscos de segurança para os doentes jovens [12].
Tipos de Nitisinone: Orfadin e Nityr
Nitisinone está disponível como medicamento genérico. É vendido sob dois nomes de marca: Orfadin e Nityr. O Orfadin foi a primeira versão do nitisinone disponível no mercado. O Nityr é bioequivalente ao Orfadin. Isto significa que ambos os medicamentos contêm a mesma substância ativa (nitisinone) e funcionam da mesma forma [8].
Dito isto, existem algumas diferenças entre os dois tipos de tratamentos nitisinone .
Orfadin vs Nityr: Qual é a diferença?
Embora tanto o Orfadin como o Nityr actuem da mesma forma e sejam prescritos para tratar a mesma doença (HT-1), diferem um do outro nos seguintes aspectos:
- Tamanho. Os comprimidos de Nityr são muito mais pequenos do que as cápsulas de Orfadin (cerca de 20% do seu tamanho) [9]. Este facto pode fazer a diferença para os doentes que têm dificuldade em engolir.
- Armazenamento. As cápsulas de Orfadin têm de ser refrigeradas (ou, se forem armazenadas à temperatura ambiente, têm de ser utilizadas no prazo de 45 dias ou eliminadas). Nityr comprimidos não requerem refrigeração [10]. Isto pode torná-los mais fáceis de tomar em viagem.
- Aprovação. Apenas o Orfadin está aprovado para o tratamento da AKU. Esta aprovação é apenas na UE [16]. No entanto, o Orfadin e o Nityr são medicamentos bioequivalentes, partilhando a mesma substância ativa e o mesmo mecanismo de ação. Isto faz com que seja apenas uma questão de tempo para que o Nityr obtenha oficialmente a mesma aprovação para o tratamento da AKU.
Custo do tratamento Nitisinone
O custo do seu tratamento em nitisinone depende de vários factores, incluindo a sua localização, o fornecedor do medicamento e qualquer cobertura de seguro a que possa ter direito.
O facto de estar a utilizar o medicamento para a sua utilização aprovada (HT-1) ou para uma utilização não autorizada (AKU) também faz diferença. Algumas companhias de seguros podem não fornecer cobertura para o uso off-label de um medicamento.
A título indicativo, estes são os preços aproximados dos comprimidos de nitisinone :
- O preço estimado do Orfadin para um mês de tratamento (com base na administração duas vezes por dia) pode variar entre cerca de 5 120 euros e 51 116 euros, consoante a dosagem necessária [12].
- Para um mês de tratamento com Nityr, os custos são estimados em cerca de 5.000 a 25.129 euros, consoante a potência necessária [13].
Nitisinone disponível gratuitamente na Índia, Paquistão, Bangladesh e Sudão
O fabricante de Nityr, Cycle Pharma, está atualmente a fornecer gratuitamente comprimidos de Nityr a doentes com AKU e HT-1 na Índia, Paquistão, Bangladesh e Sudão. Isto é possível graças a uma parceria estabelecida entre Everyone.org e Cycle Pharma em 2022.
Note-se que podem aplicar-se algumas condições. Se reside na Índia, Paquistão, Bangladesh ou Sudão, envie um pedido para Nityr através da página abaixo para saber se tem direito a receber gratuitamente os comprimidos nitisinone (Nityr).

NOTA: Infelizmente, atualmente não podemos fazer entregas no Sudão. No entanto, podemos disponibilizar Nityr para recolha numa das nossas farmácias parceiras no Luxemburgo, Alemanha ou Países Baixos. Contacte-nos para mais informações.
O futuro dos tratamentos das doenças raras
Embora atualmente apenas 5% das doenças raras tenham um tratamento, vemos este panorama médico a mudar todos os dias. Nitisinone é apenas um exemplo. Mal podemos esperar para partilhar mais consigo.
Referências:
- Sintomas e tratamento da tirosinemia. UPMC Children's Hospital of Pittsburgh, Acedido em 2 de agosto de 2023.
- Tirosinemia tipo 1 - Sobre a doença. Centro de Informação sobre Doenças Genéticas e Raras, Acedido em 2 de agosto de 2023.
- Feillet, François. Adesão ao tratamento na Tirosinemia Hereditária Tipo 1 (HT1): A Mixed-Method Investigation into the Beliefs, Attitudes and Behaviour of Adolescent Patients, Their Families and Their Health-Care Team (Uma investigação de método misto sobre as crenças, atitudes e comportamentos dos doentes adolescentes, das suas famílias e da sua equipa de cuidados de saúde). NCBI, 12 de setembro de 2014.
- Braekeleer, M., e J. Larochelle. Epidemiologia genética da tirosinemia hereditária no Quebec e em Saguenay-Lac-St-Jean. NCBI, Acedido em 2 de agosto de 2023.
- Uma comparação das frequências de doenças e genes de erros inatos do metabolismo entre diferentes grupos étnicos em West Midlands, Reino Unido. Journal of Medical Genetics, maio de 1998.
-
Das, Martin. Utilidade clínica de nitisinone para o tratamento da tirosinemia hereditária tipo 1 (HT-1). NCBI, 24 de julho de 2017.
-
Nitisinone Oral: Usos, efeitos secundários, interacções, imagens, avisos e dosagem. WebMD, Acedido em 2 de agosto de 2023.
-
Nityr. Agência Europeia de Medicamentos, Acedido em 2 de agosto de 2023.
- O que é Nityr?. Nityr, Acedido em 2 de agosto de 2023.
- ID de referência: 4130114. Accessdata.fda.gov, Acedido em 2 de agosto de 2023.
- Adequação de nitisinone para a gestão da alcaptonúria. NCBI, Acedido em 2 de agosto de 2023.
- Preços de Orfadin, cupões, copagamento e assistência ao paciente. Drugs.com, Acedido em 2 de agosto de 2023.
- Nityr Preços, cupões, copagamento e assistência ao paciente. Drugs.com, Acedido em 2 de agosto de 2023.
- Tyrosinemia type I. Myriad Genetics, Acedido em 2 de agosto de 2023.
- Sucesso no DevelopAKUre: aprovação do Orfadin para o tratamento de doentes com AKU. Investigação e inovação, 26 de outubro de 2020, Acedido a 2 de agosto de 2023.